Introduction
Congenital adrenal hyperplasia (CAH) is an autosomal recessive genetic disorder arising from a lack of one of enzymes indispensable for cortisol biosynthesis. 17 α -hydroxylase deficiency (17 α-OHD) is an infrequent variant of CAH, resulting from mutations in CYP17A1 gene. It represents approximatively 1% of the total CAH cases. Deficiency of the enzyme leads to compromised cortisol and sex steroid production, provoking a compensatory increase in adrenocorticotropic hormone (ACTH) and gonadotropin levels.[1] Clinically, 17 α-OHD is typically marked by hypertension, hypokalemia, sexual infantilism and delayed puberty .[2]
In this paper, we present the case of two siblings who had 17 α-OHD with different clinical presentations.
Case presentation
Case 1:
A 36-year-old woman with hypertension since age 30 was referred for primary amenorrhea and incidentally discovered bilateral adrenal hyperplasia. She was born to consanguineous parents. Her older sister, aged 42, had also been investigated for primary amenorrhea at age 17 and was treated with hormone replacement therapy, leading to temporary menstruation, which ceased 15 years after stopping treatment.
In 2023, at age 34, a CT colonography for chronic constipation incidentally revealed bilateral adrenal hyperplasia. On examination, her BMI was 33 kg/m², and blood pressure was 120/70 mmHg under calcium-channel blocker. No skin hyperpigmentation was noted. She had Tanner stage 3 breast development and stage 1 pubic hair (S3P1). External genitalia were female and infantile, with a hymenal ring and vaginal dimples.
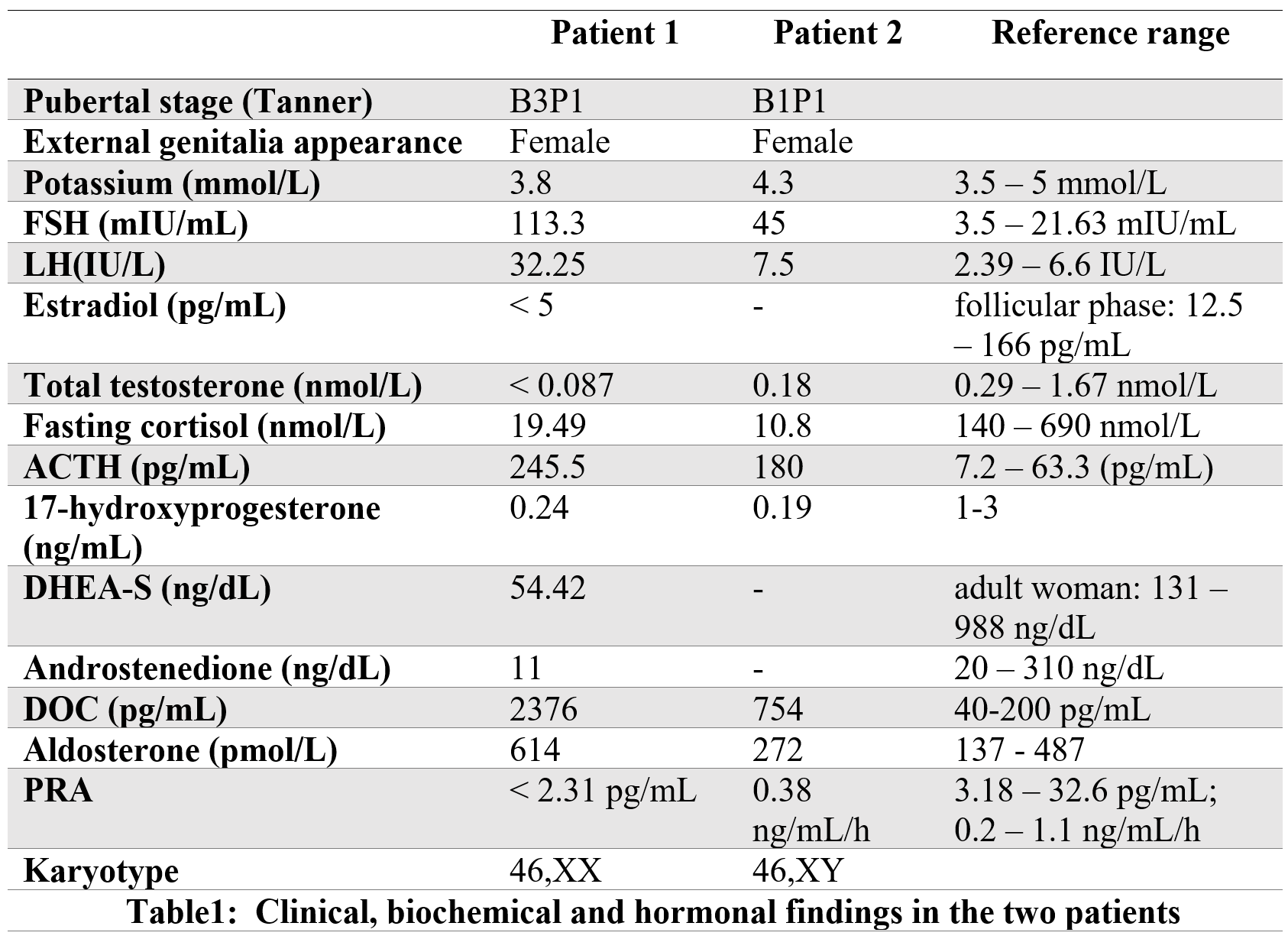
Lab tests showed normal potassium. Adrenal insufficiency was confirmed by low cortisol and elevated ACTH. Hypergonadotropic hypogonadism was indicated by elevated FSH and LH, with low estrogen and testosterone. Hormonal profile also showed high 11-deoxycorticosterone (DOC), renin, and aldosterone, and low levels of 17-hydroxyprogesterone, androstenedione, and DHEA-S (Table 1). Pelvic ultrasound showed an immature uterus without visible ovaries; MRI confirmed a hypoplastic uterus and ovaries. Karyotype was 46,XX.
Based on clinical, biochemical, and imaging findings, the diagnosis of 17α-hydroxylase deficiency (17α-OHD) was made. Bone mineral density revealed osteoporosis. She was treated with estradiol, progesterone, risedronate, hydrocortisone, and amlodipine. Sexual development progressed (Tanner S5P5), and she began regular menstrual cycles.
Case 2:
The older sister was first seen at age 15 (in 1996) for delayed puberty. She was 159 cm tall (−1 SDS), weighed 60 kg (+1 SDS), and had normal blood pressure (120/70 mmHg). She presented with female sexual infantilism (Tanner stage 1), with no menarche. Potassium was normal (4.3 mmol/L).
Hormonal evaluation showed elevated FSH and LH (hypergonadotropic hypogonadism), low cortisol, and high ACTH, indicating adrenal insufficiency (Table 1).
Pelvic CT revealed bilateral adrenal hyperplasia and absence of uterus and ovaries. Karyotype was 46,XY, raising the suspicion of a disorder of sex development associated with adrenal insufficiency. The patient had female gender identity and phenotype despite 46,XY karyotype.
Further testing showed low testosterone and 17-hydroxyprogesterone, high DOC, and normal renin and aldosterone. Based on these findings, 17α-OHD was diagnosed. Hydrocortisone therapy was initiated.
Due to the absence of palpable gonads and internal genitalia, an intraperitoneal exploration was performed. No Müllerian structures were identified. Pelvic exam showed a 2 cm vaginal depth, no cervix or uterus. Two small gonads (1 cm and 0.5 cm) and rudimentary mesonephric remnants were found near the pelvic brim. Histology confirmed prepubertal testes. The patient underwent bilateral cryptorchidectomy and was started on estrogen therapy.
Bone mineral density testing confirmed osteoporosis, and risedronate was prescribed. Blood pressure remained normal on follow-up. She continued estrogen therapy with good tolerance. Vaginoplasty was performed. She later married and reported no difficulties in social or sexual life.
Discussion
The earliest known case of 17α-OHD was documented by Biglieri et al. in 1996, presenting a 35-year-old female patient, with hypertension, hypokalemia, and sexual infantilism.[3] Over 500 cases have been registered up until now.[4] 17 α-OHD is caused by a mutation in the cytochrome P450c17 encoded by the CYP17A1 gene. Expressed in adrenal cortex end the gonads, P450c17 catalysis both steroid 17α-hydroxylase and 17,20-lyase activities and serves as a critical component in the biosynthesis of cortisol and adrenal steroids.[5] The initial reaction involves 17α-hydroxylase activity, leading to the conversion of pregnenolone and progesterone into their respective 17α-hydroxylated forms, namely, 17α-hydroxypregnenolone and 17α-hydroxyprogesterone. The second reaction depends on 17,20-lyase activity, catalyzing the transformation of these newly formed 17α-hydroxysteroids into dihydroepiandrosterone and androstenedione. Consequently, loss of function of 17α-hydroxylase could explain the decreased level of the 17-hydroxyprogesterone in the two cases.
Since both adrenal and gonadal steroid synthesis is impaired in patients with 17 α-OHD, clinically primary amenorrhea and sexual infantilism are seen in girls (46, XX) and disorders of sex development are seen in boys (46, XY). The two siblings in our presentation had female phenotype. Yet, during the karyotype analysis, it was demonstrated that the first case had a 46, XX karyotype, while the second case had 46, XY.
Our first case presented female infantile external genitalia with a partial breast development which is probably due to the ethinylestradiol treatment that she had received in the past.
In individuals with 46, XY, Müllerian ducts (including fallopian tubes, uterus, and the upper third of the vagina) are absent due to normal secretion of anti-Müllerian hormone during the embryonic stage. Disrupted gonadal and adrenal androgen production leads to hypoplastic testes in terms of internal genitalia, while the external genitalia appear as infantile female, as described in our second case, or ambiguous, in alignment with the extent of the enzymatic obstruction.[6] In facts, studies have shown that a minimum of 25% of normal enzymatic activity is required for adequate fetal masculinization. [7] In response to decreased cortisol synthesis, there is a compensatory overproduction of ACTH, leading to bilateral adrenocortical hyperplasia as documented in our cases. This excess ACTH also results in an excessive amount of DOC production, revealing hypertension and hypokalemia. Patients are typically diagnosed during adolescence because of delayed puberty, hypertension, and hypokalemia.[8] These signs were not found together in our two patients. In fact, blood potassium level was normal in the 36-year-old patient who had hypertension. Her sister had been always normotensive. Indeed, around 10–15% of patients identified with 17 α-OHD exhibit normal blood pressure and potassium levels when diagnosed. This variability can be explained by the differential sensitivity of various cortisol precursors to target tissues exhibiting mineralocorticoid activity. Based on previous studies, the majority of patients have low or within normal ranges of aldosterone levels. This is attributed to the renin suppression secondary of sodium retention due to excessive DOC production 2. Our second patient who presented normal level of aldosterone, supports this hypothesis. Nonetheless, similar to her sister, elevated aldosterone levels have been described in some patients. The prevailing theory is that increased aldosterone levels in patients may be attributed to a more pronounced enzyme’s deficiency, resulting in heightened activity of corticosterone methyl oxidase in fasciculate cells. Thus, aldosterone derived from corticosterone will be increased, downregulating, as a result, renin production.[9] Neither of our cases displayed clinical symptoms related to adrenal insufficiency. This can be attributed to the partial glucocorticoid activity of corticosterone.
Management strategies for 17α-OHD are mainly based on glucocorticoid supplementation and adrenal steroid hormone replacement. Hydrocortisone helps restore negative feedback on the corticotropic axis, leading to a reduction in ACTH levels and a decrease in the excessive production of mineralocorticoids, controlling hypertension. When the diagnosis is established at a later stage in life, as seen in our first case, prolonged hypertension may render this therapy ineffective. The association to a mineralocorticoid antagonist, such as spironolactone, and calcium channel blockers has been useful to achieve better blood pressure management.[10]
Regarding the hypogonadism, Steroid hormone replacement is crucial not only to preserve female sexual characteristics, but also to improve the cardiometabolic profile and prevent bone mass loss 10. In individuals with a karyotype of 46, XX, estrogen and progestin replacement therapy can be employed to initiate the menstrual cycle. A risedronate treatment can be started if osteoporosis is already in place, as illustrated in our cases. Prior to initiating treatment, medical professionals should engage in discussions with patients or their parents for pediatric or adolescent patients as it is the case of our patient (case 2) regarding the treatment's benefits, potential effects and consequences. Physicians should also honor patients' choices regarding treatment, including gender preferences. In light of our patient's female phenotype and gender, bilateral gonadectomy was conducted upon the observation of cryptorchidism to prevent gonadal malignancy.
Addressing our study's limitations, it is essential to highlight the fundamental role of genetics in the diagnosis of this disorder. Regrettably, our hospital does not have access to these tests.
Conclusion
In conclusion, we've encapsulated two different presentations of 17 α-OHD in two siblings, with 46, XX and 46, XY karyotypes. Because, this disease is relatively rare, it should be considered in adolescence as well as in adulthood. Early identification of the disorder is essential to establish a well-structured and personalized long-term management strategy, alleviating its complications notably hypertension and osteoporosis, as well as improving patients’ quality of life.
Consent and ethics
Written informed consent was obtained from both patients for publication of this case report and accompanying clinical details.
References
- Ammar R, Ramadan A. Incidental diagnosis of 17 alpha-hydroxylase deficiency: a case report. Oxf Med Case Reports. 2020;2020(12):omaa108. doi:10.1093/omcr/omaa108
- Yanase T, Simpson ER, Waterman MR. 17 alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991;12(1):91-108. doi:10.1210/edrv-12-1-91
- Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man. J Clin Invest. 1966;45(12):1946-1954. doi:10.1172/JCI105499
- Wang M, Wang H, Zhao H, et al. Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese Han population. Clin Hypertens. 2019;25:23. doi:10.1186/s40885-019-0128-6
- Unal E, Yıldırım R, Taş FF, Tekin S, Ceylaner S, Haspolat YK. A rare cause of delayed puberty in two cases with 46,XX and 46,XY karyotype: 17 α-hydroxylase deficiency due to a novel variant in CYP17A1 gene. Gynecol Endocrinol. 2020;36(8):739-742. doi:10.1080/09513590.2019.1707798
- Jiang JF, Deng Y, Xue W, Wang YF, Tian QJ, Sun AJ. Surgical Therapy of 17α-hydroxylase Deficiency in 30 Patients. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2016;38(5):559-562. doi:10.3881/j.issn.1000-503X.2016.05.012
- New MI. Male pseudohermaphroditism due to 17 alpha-hydroxylase deficiency. J Clin Invest. 1970 Oct;49(10):1930-41. doi: 10.1172/JCI106412.
- Bee YM, Manju C, Papari-Zareei M, Auchus RJ. Phenotypic variation in a Chinese family with 46,XY and 46,XX 17α-hydroxylase deficiency. Gynecol Endocrinol. 2012;28(4):322-325. doi:10.3109/09513590.2011.631625
- Yamakita N, Murase H, Yasuda K, Noritake N, Mercado-Asis LB, Miura K. Possible hyperaldosteronism and discrepancy in enzyme activity deficiency in adrenal and gonadal glands in Japanese patients with 17 alpha-hydroxylase deficiency. Endocrinol Jpn. 1989;36(4):515-536. doi:10.1507/endocrj1954.36.515
- Bouça B, Cascão M, Fiúza P, Amaral S, Bogalho P, Silva-Nunes J. Diagnosis of 17-alpha hydroxylase deficiency performed late in life in a patient with a 46,XY karyotype. Endocrinol Diabetes Metab Case Rep. 2023;2023(2):22-0338. doi:10.1530/EDM-22-0338